[Computational Sciences]Basic Calculation Using PHASE0
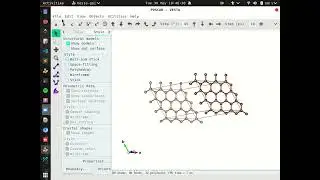
Calculating Crystal System using PHASE0
1. Preparing input files SCF or Self Consistent Field Calculation:
filenames.data : declaration of files that we need to calculate
input.data : cyrstal system, accuracy configuration, control calculation etc.
pseudopotential.pp : density functional theory or approximate the potential of the atoms.
Each atoms has a their pseudopotential.
Silicon : silicon pseudopotential
Silicon Carbide : silicon and carbon pseudopotential
2. Execute phase:
mpirun -np 2 phase
3. Check the progress of your calculation:
cat jobstatus.data : job status in your calculation.
cat output000
cat nfefn.data : total energy
cat continue.data : continue 2 (convergence)
nfchgt.data : it is needed for the bandstructure and density of states
▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬
Hello there 🤓🤓🤓
I am Zohan Syah Fatomi
I am a Postgraduate Student of Physics at Universitas Gadjah Mada. I have a great interest in Mathematics, Physics, Codes (Python), Numerical Method and Universe. Sometimes Writing a Book too ... 👨🎓
▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬
Pls help us support this channel
👉Subscribe: https://bit.ly/2Db1J0O😊
Semoga bisa bermanfaat
▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬▬
On this page of the site you can watch the video online [Computational Sciences]Basic Calculation Using PHASE0 with a duration of hours minute second in good quality, which was uploaded by the user Zohan Syah Fatomi 02 October 2022, share the link with friends and acquaintances, this video has already been watched 166 times on youtube and it was liked by 3 viewers. Enjoy your viewing!







![[FREE FOR PROFIT] Future × Metro Boomin Type Beat](https://image.4k-video.ru/id-video/Kv1JgAWbUrc)

![Python Dasar 5: Tipe Data Variabel di Python [BAHASA INDONESIA]](https://image.4k-video.ru/id-video/otCg2Vp-wsE)
![Python Dasar 4: Variable di Python [BAHASA INDONESIA]](https://image.4k-video.ru/id-video/Vx8f0R38MoI)
![Python Dasar 3: Tutorial GOOGLE COLAB & JUPYTER NOTEBOOK [BAHASA INDONESIA]](https://image.4k-video.ru/id-video/K5vIhXhp7uY)
![Python Dasar 2: Apa itu Programming ??? [BAHASA INDONESIA]](https://image.4k-video.ru/id-video/_WRHwgKr37U)
![Python Dasar 1: Menginstal Python 3.11.4 di Ubuntu 22.04 LTS [WORKED] [BAHASA INDONESIA]](https://image.4k-video.ru/id-video/3dr5-eUA_7o)
![[Computational Sciences]Basic Calculation Using PHASE0](https://image.4k-video.ru/id-video/v2iVtZijyTY)
![[Computational Science] Download and Installing PHASE0 DFT Code](https://image.4k-video.ru/id-video/EOhtXnGdPJw)
![How to CONVERT PNG to JPG Without SOFTWARE in LINUX?! [SOLVED]](https://image.4k-video.ru/id-video/KVpDjr9JRps)
![How To Install KeyBase in Linux 20.04 [SOLVED]](https://image.4k-video.ru/id-video/IE6CVapFtlc)